
Assisting families and individuals in the bleeding community
@jordanhannafoundation
Our application for NPO registration is underway
ABOUT JHF
As humans – we want to leave a legacy and contribute in some way to society. We try donate to organizations where we can and do our part – but we, have fortunately been afforded the opportunity to create a Foundation that is close to home and that has affected us personally.
When you say the word Haemophilia – we more often than not – receive an unsure look as most people have never heard of Haemophilia and what it means. For this reason, amongst others – we have started the Jordan Hanna Foundation.
This is our story…
On the 3rd December 2018, a 3 day old – Jordan – underwent a routine circumcision. We went home soon after the procedure – but 4 hours later we rushed him back – after his blood soaked nappy alerted us – that something was very wrong.
It was then that we found out that our little miracle baby boy – that we waited 8 years for – after countless fertility hurdles and one successful IVF transfer – had Haemophilia A – a genetic blood disorder.
This meant his Factor 8 gene had mutated and his blood would not clot like normal.
It never crossed our minds to test me as a Haemophilia carrier when doing all the numerous genetic tests while pregnant – as the condition has not resurfaced since my Grandfather had it.
The Haemophilia gene is carried by the mother’s one X chromosome and a boy – has a 50% chance of receiving the defective X gene and therefore would experience the bleeding condition.
Life as we knew it changed in a matter of minutes that day. Everything that we thought our new life as a family would be, was now masked with a veil of extra padding… constant eagle eyes… attempting to avoid every knock and bump as possible – all to prevent an internal bleeding episode.
We started noticing bruising around his ribs when we picked him up a certain way – fingerprint bruises almost – so we had to change the way we picked him up and the way we handled this new baby.
When he knocked his head – as babies do – we would have to take him to the nearest Haemophilia hospital – Unitas in Centurion – to infuse Jordan with Factor 8 replacement – so that his blood would clot and the bump would not turn into something worse – or even a brain bleed.
This exercise was traumatic for both parties – parents’ and child – with babies it is sometimes difficult to find a vein to administer the meds and eventually they would have to wrap him up and find a vein in his foot. (One of the most painful parts of the body).
After 18 months of trips to the hospital for infusions – sometimes it was a precautionary infusion and sometimes it was VERY necessary.
The last day of vein access transfusion – was after he bumped his head – when the nurses tried 6 times to find a vein and the doctor advised the nurse to try the vein in his head.
We then decided it was necessary to insert a portacath. A small device that is surgically implanted into Jordan’s chest – and a small tube is inserted into a main artery.
Cancer patients use this method of access when doing chemotherapy.
His medication would have to be administered 3 times per week – this is called prophylaxis – so his Factor levels would be maintained and the chances of him spontaneously bleeding or having a serious internal joint bleed would be minimal.
Jordan’s current Factor level is between 1% and 2% without receiving Factor 8 replacement – making him a Severe to Moderate bleeder. A normal level is above 50%
This rare chronic condition is a life long condition and there is currently no cure.
Jordan is healthy and thriving and is a busy normal little boy. He does not know he is different to anyone around him – except that he has a “shark bite” (the name we gave his port after the implant) and that his “muti” (Factor) makes his “eina’s” better and his bleeding stop.
He knows no limits and we have tried to raise him without putting him in a bubble.
To us and him… he is normal – and that’s how he needs to live – except he just has his “muti” every 3 days.
We are truly and whole heartedly grateful that we are living in a time that Haemophilia patients have better access to Factor and there is more knowledge & awareness (but there is still not enough) around this condition.
50 years ago patients truly suffered and are still suffering from the impact of not receiving or even knowing about prophylaxis treatment.
To prevent internal bleeding – especially joint bleeds is the number one priority – the more bleeds that occur in a joint – will eventually lead to a target joint and the deterioration of that joint. Therefore the patient suffers tremendous pain and stiffness – eventually a joint replacement is necessary. Some children by the age of 13 need joint replacements from all the bleeds in a specific joint.
We are living in a country that considers Haemophilia a chronic condition and we are so grateful we have medical aid that covers the monthly spend – for now this is nearly R32 000.00 per month for his medication and consumables. This will increase as he gets older as he will need more Factor to match is body weight.
Our government does provide Factor at certain hospitals for free but this is not always available when a patient needs it.
Our aim for the Jordan Hanna Foundation is to bring more awareness about this rare condition and to raise funds to help those families that can’t get to the life saving medications or require assistance.
WHAT IS HAEMOPHILIA?
Hemophilia or Haemophilia is an hereditary condition. This means that it is passed on from mother to child at the time of conception.
The blood of a person with haemophilia does not clot normally. He does not bleed more profusely or more quickly than other people; however, he bleeds for a longer time.
Many people believe that haemophiliacs bleed a lot from minor cuts. This is a myth. External wounds are usually not serious. Far more important is internal bleeding (hemorrhaging). These haemorrhages are in joints, especially knees, ankles and elbows; and into tissues and muscles. When bleeding occurs in a vital organ, especially the brain, a haemophiliac’s life is in danger.
Haemophilia A, or Classical Haemophilia, is the most common form, and is caused by having reduced levels of factor VIII (8). Haemophilia B, or Christmas Disease, is caused by having reduced levels of factor IX (9).
What causes the bleeding?
Bleeding is often caused by minor injury – a bump or a slight twist of a joint. However, many haemorrhages, especially among severe haemophiliacs, happen for no apparent reason. This is even truer in joints that have bled often in the past. The more a joint has bled, the easier it bleeds again with no external cause.
Even haemorrhages in the brain often have no apparent cause. Brain haemorrhages are the leading cause of death from bleeding in haemophilia. Therefore it is important to recognise the symptoms of a brain haemorrhage very quickly.
How Haemophilia is inherited
Haemophilia is an inherited condition and occurs in families; however, in 1/3 of cases it appears in families with no previous history of the disorder. The genetic alteration causing haemophilia is passed down from parent to child through generations. Men with haemophilia will pass the altered gene on to their daughters but not their sons. Women who carry the altered gene can pass it on to their sons and daughters. Sons with the gene will have haemophilia. Most women and girls who carry the gene do not have bleeding symptoms. However, others may have a bleeding tendency, especially if they have have low factor levels. These women and girls will have haemophilia and need to receive treatment.
The diagrams below may assist in understanding this. The red males are those with haemophilia; the red and blue females carry the gene – they have X chromosome with the genetic alteration and one unaltered X chromosome.
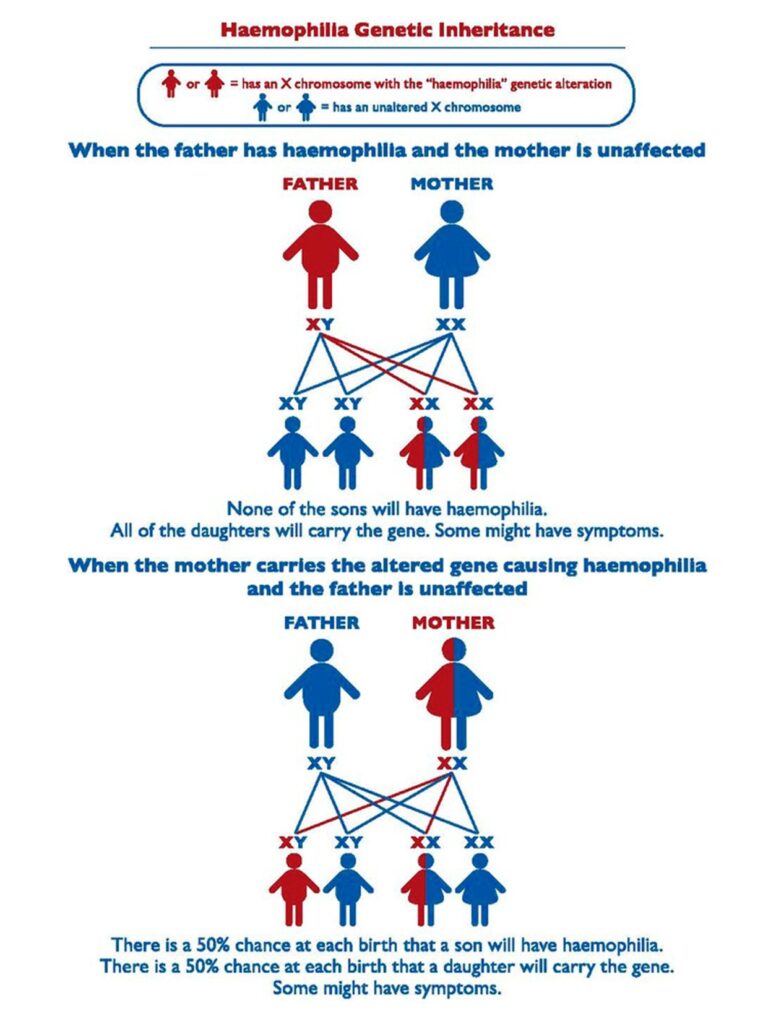
Who is affected by Haemophilia?
Haemophilia affects people of all races, colours and ethnic origins.
The most severe forms of haemophilia affect almost only males. Females can be seriously affected only if the father is a haemophiliac and the mother is a carrier, or in the case of X-inactivation when a woman’s normal X-chromosome is inactive in the production of factor VIII or IX. These cases are extremely rare.
However, many women who are carriers have symptoms of mild haemophilia. We are only now fully recognising the importance of bleeding in carriers and the degree to which these symptoms affect a woman’s quality of life.
As haemophilia is an hereditary disorder, people are affected at birth. This means that children can have haemophilia. In fact, haemophilia is often diagnosed in the first year of life.
How serious is Haemophilia?
Without proper treatment, haemophilia is crippling and often fatal. With modern treatment, most people with haemophilia can lead full, active lives. Haemophilia is classified as severe, moderate or mild.

Severe haemophiliacs with less than 1% of the normal level of factor VIII or IX in the blood have haemorrhages several times a month. The bleeding is often the result of a minor bump or twist. Sometimes, there is often no apparent cause for the bleeding. Moderate haemophiliacs bleed less often. Their haemorrhages are often the result of minor trauma, such as a sports injury. Mild haemophiliacs have even fewer haemorrhages. They may be aware of their bleeding problem only in the case of surgery, a tooth extraction or a serious injury. Women with mild haemophilia may bleed more during menstruation (periods).
MYTHS & FACTS
Myth: If a person with hemophilia gets a cut, he’ll bleed to death.
Highly unlikely. People with hemophilia do not bleed any faster or harder than those without the disease, they simply bleed for longer. Even so, superficial cuts or abrasions are generally not a cause for concern and a bandaid will suffice in most cases. For joint and muscle bleeds, apply usual first aid treatment including rest, ice, compression and elevation (or R.I.C.E). The real concern for serious bleeding is if internal organs and/or deep tissues, muscles and joints are hurt. After any injury, if in doubt, contact your treatment team.
Myth: Hemophilia affects only boys.
While most people assume hemophilia only affects boys, occasionally girls who are “silent carriers” can suffer bleeding symptoms as well. Though extremely rare, a daughter who is born to a father with hemophilia and a mother who is a carrier can actually inherit the severe form of the disease.
Myth: Everyone with hemophilia has the same level of deficiency.
Nope. “Depending upon the specific genetic change or mutation in a given family, the severity of the deficiency or level of clotting factor in the blood varies from a severe deficiency to moderate to more mild forms of the disease,” says Dr. Fahner.
Myth: Family members can have different clinical severities of hemophilia.
No. Given that the same gene defect is passed down through the family, all members will have the same level of severity. Some may seem like they have milder or more severe cases depending on how active and/or accident-prone they are, but unless there has been a genetic mutation all family members will be identical in the severity they express. There are caveats to this, however: other factors may influence the severity seen in individuals in the same family, including the presence of inhibitors or other bleeding and coagulation disorders. In addition, if one family member gets an inhibitor, it raises the risk of other siblings getting one as well — although this risk isn’t absolute.
Myth: All forms of hemophilia involve a deficiency in clotting factor VIII.
Not true. Hemophilia A (also known as Classical Hemophilia and Factor VIII Deficiency Hemophilia) is the most common and is a deficiency in clotting factor VIII. However, Hemophilia B (aka Christmas Disease and Factor IX Deficiency Hemophilia) is—you guessed it—a deficiency in factor IX. Hemophilia C occurs from a lack in factor XI.
Myth: Patients with hemophilia always have a family history of the disease.
“A very important false,” says Dr. Fahner. “The gene defect responsible for hemophilia has one of the highest rates of spontaneous brand new mutations of any human genetic disorder. Some researchers estimate that as many as one-third of the cases of hemophilia are new mutations with no family history of the disease.”
Myth: Everyone with hemophilia will eventually get bad joints.
While this used to be very often the case, the good news is that, with safer and more readily available clotting factor concentrates, many people with hemophilia today will be able to prevent long-term joint injury and crippling arthritic changes. Key to this is patients receiving primary (preventative) prophylaxis where they are given a dose of factor to actually prevent a bleed rather than treating it once it has begun, starting before any joint damage has occurred.
Myth: It’s ok to wait until one sees something and really knows it’s a bleed before acting.
This is a big no in any situation. “Act immediately because especially in brain trauma it can be life and death,” says Dr. Mills. When in doubt, treat and then get it checked out.
Myth: Persons with Hemophilia can’t play sports or have a “normal life”
Those individuals with Hemophilia and other bleeding disorders are actually encouraged to play team sports and get exercise like everyone else. Activity is important for building muscle strength, promoting joint health and a healthy weight, all of which are essential for everyone, including someone with a bleeding disorder.
Choosing the right sports and activities for an individual is best done in conjunction with the health care team. With current therapies used to treat and prevent bleeding episodes and the establishment of specialized treatment centers, like the Hemostasis and Thrombosis Center at Rhode Island Hospital, people with bleeding disorders are living long and healthy lives.
JHF EVENT'S
CONTACT US
E-Mail: info@jhfoundation.co.za
Mobile: 082 487 4773
Donate with SnapScan
SnapScan is an app that lets you pay with your phone quickly, easily, and safely. The SnapScan app uses your phone’s camera to scan SAHF’s unique QR code (pictured left)